核酸とタンパク質の結合機構に関する研究
平成18年3月
お茶の水女子大学理学部
今野 美智子
研究目的
核酸のなかでもRNAの塩基を選択し結合するアミノアシルtRNA合成酵素(aaRS)の構造を研究する。この酵素群における塩基選択的結合の機構とその構造的共通性の解明を進める。同時に、基本的な機能を保持しつつ、進化の過程で機能的多様化を獲得する分子生物学的タンパク質進化との関連性を含めた相互比較を行う。
tRNAは、すべてに共通な3次構造を持ち、tRNAはアンチコドンの塩基の違いにより多様な構造をもつaaRSによって厳密に識別される。コドンの1番目と2番目は、各アミノ酸に対して1種類であることから、tRNAのアンチコドンの2番目と3番目の塩基は、aaRSと水素結合のような強い相互作用を形成して結合し識別されるが、特に、中心にある2番目のアンチコドンは、供与体、受容体として2つの水素結合が形成され、3番目は、1つの水素結合の形成があると予想される。一方、3番目のコドンは多様性を持つことから、アンチコドンの1番目は、aaRSと弱い相互作用と立体障害による排除の機構も働く。本研究は、tRNAのアンチコドンの3個の各塩基におけるそれぞれ独立な相互作用の機構を調べ、すべてのtRNAに共通に働くかどうか解明すること目的とし、アンチコドン結合ドメインが同じヘリックス-バンドル構造を持つクラスIaのaaRS酵素を中心に研究を進めている。コドンの使用頻度の高いものは、それに対応するアンチコドンのtRNAの反応活性が高くなる。コドンの使用頻度の変化によるアンチコドンの塩基の変化に対応して、その塩基を識別するサイトは同じであるが、認識に関与するアミノ酸残基が置換されると考えられ、それらを比較することにより関与するアミノ酸残基の情報が得られる。真性細菌である大腸菌、Thermus thermophilus (T.t)、真核生物の酵母菌、古細菌であるPyrococcus horikoshii (P.h.)のコドンの使用頻度を比較し、その使用頻度に大きな差のあるもの、その中でも、真性細菌と古細菌で使用頻度に顕著の違いがあるArg、Ile、GlyにおいてT.t. aaRSの結晶構造は報告されており、それとの比較において古細菌、主にP.h.由来aaRSを取り上げた。
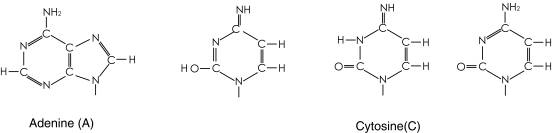
図1
表1に示すように、Ile、Met、Valは、2番目のコドンがUra、対応するアンチコドンがAdeである。他方、Trp、Arg、Glyは、2番目のコドンがGua、アンチコドンではCytである。この2番目のアンチコドンAdeとCytの識別において、図1に示すようにAdeは-NH2-C6=N1-で供与体、受容体として2つの水素結合を形成すると思われる。Cytは主に3つの共鳴構造を持つが、疎水的環境では中央の共鳴構造の存在率が高くなる。この共鳴構造の-N3H-C2=O-と供与体、受容体として2つの水素結合を形成することによりAdeとの識別の違いが生ずると思われる。
表1
|
|
codon |
anticodon |
|
codon |
anticodon |
|
|
|
|
|
Trp |
UGG |
CCA |
|
|
Arg |
CGU CGC CGA CGG AGA AGG |
ICG CCG UCU CCU |
||||
Ile |
AUU AUC AUA |
AAU GAU UAU |
||||
Met |
AUG |
CAU |
||||
|
Val |
GUU GUC GUA GUG |
UAC CAC |
Gly |
AAU GGC GGA GGG |
GCC UCC CCC |
赤:構造解析されたtRNAのアンチコドン
表2 Argのコドンの使用頻度とアンチコドン
|
codon |
E.coliUsage
anticodon |
T. thermophilusUsage
anticodon |
Yeast Usage
anticodon |
P. horikoshiiUsage
anticodon |
||||
|
CGU CGC CGA CGG |
28 21 3 4 |
ICG |
1 3 0 33 |
CCG |
6 3 3 2 |
ICG CCG |
1 1 1 0 |
|
|
AGA AGG |
1 1 |
|
1 14 |
CCU |
21 9 |
UCU CCU |
19 34 |
UCU CCU |
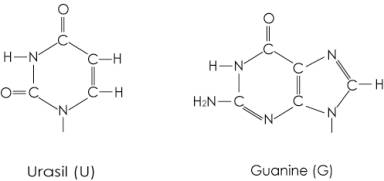
表2に示すように、Argの主な使用頻度のコドンは、大腸菌ではCGU/CGC/CGA/CGGであり、T.t.あるいは酵母菌でも広がっているが、P.h.では、AGA/AGGが98%で、AGGが最も高く選択の機構がより反映される。P.h.の主なtRNAArgのアンチコドンは、CCUとUCUであり、酵母菌のそれは、ICGとUCUである。図2に示すように3番目のアンチコドンGua と Uraに共通な相互作用として-C=O-NH-との水素結合が可能である。1番目のアンチコドンCytとUraに共通な部位として、Cの共鳴構造が-NH2-C=N-C=O-から-NH=C-NH-C=O-に移行するとCytのNHとUraのC=Oとイノシン(I)のC=Oの孤立電子対を共通に供与することができる。これらの共通部分がタンパク質と相互作用すると予測される。
図2 tRNAArgのアンチコドン
P.h. 酵母菌
CCU ICG
UCU UCU
3番目のアンチコドン
1番目のアンチコドン
更に、ArgのアンチコドンCCUは、MetのアンチコドンCAUに対し、2番目のみが異なる。また、Ileのコドンの使用頻度は、P.h.において3種類のコドンの中でAUAが高く、Metのアンチコドンと1番目が異なるのみである。他方、T.t.のIleのコドンはAUCが極端に高い。クラスIcであるTrpRSもTrpの2番目のアンチコドンがArgと同じCytであり、クラスIIに属するGlyRSにおいても、Glyの2番目のアンチコドンがCytであり、Cytの識別に対し、クラスIと同じ結合機構が使われるか検証することの意味においてもGlyRSを研究対象とした。その他、クラスIIのaaRSの触媒ドメインと同じトポロジーをもち、ATPを補酵素としMg2+イオンの存在下で反応が促進されるBiotin Protein Ligase (BirA)についても反応機構の共通性を検証するために構造解析を進めた。
平成17年度の研究成果
1) P. h. ArgRSの結晶構造解析の結果
P. h. ArgRS、tRNA(CCU)とAMP-PNPの複合体及びP. h.
ArgRSとtRNA(CCU)の複合体の構造解析に成功したので、ArgRSを中心に報告する。超高度高熱菌タンパク質は溶解度が高く、種々の結晶化を試み、単独のタンパク質の結晶が得られたが、低分解能であった。複合体の形成が、溶解度を下げることから、tRNAとの複合体の結晶化を試みた。また、ArgRSはN末端側に付加的なドメインをもつ。このN末端ドメインの配向がTt ArgRSと酵母菌ArgRSで異なることより、tRNAとの複合体がパッキングを変える可能性を考えた。6種類のtRNAのなかで最も使用頻度の高いtRNAArg(CCU)の大量発現系を確立し精製を行った。最終的に、P.h. ArgRSとtRNAArgの複合体およびAMP-PNPを含む複合体の解析に成功した。反射強度測定は、Spring8とPFのビームラインを用い100KにおいてCCDデイテクターで収集した。まず、ArgRS、tRNAとAMP-PNPの複合体の構造解析を、分子置換法でT.t.ArgRS(PDB:1IQ0)を分子モデルとしてMOLREPを用いて行った。tRNAの分子のモデルは、酵母菌ArgRSとtRNAArg複合体(PDB:1F7V)のtRNAArgを用い、酵母菌ArgRSをT.t.ArgRSに重ねてtRNAの位置を求めた。この構造で精密化しR=40%まで収束した。P.h.のArgRSの約100残基のN末端ドメインについては、タンパク質のコア部分に対する相対的位置がT.t.と酵母菌のそれらに対し大きくずれており、また、tRNAについてはL字型の曲がりが若干異なっており、これらの部分における電子密度図がぼやけていた。プログラムOによりこれらの領域の構築部分を広げながら繰り返し分子構築とプログラムCNSを用いた精密化を行うことにより、全体のモデルが構築された。最終的に、ArgRS、tRNAとAMP-PNP の複合体では、分解能2.0 ÅでRfactor =0.215 (Rfree = 0.259),
ArgRSとtRNAの複合体では、分解能2.3
ÅでRfactor =0.206 (Rfree = 0.266)である。P.h.のArgRS、tRNAのリボンモデルを図3に示す。
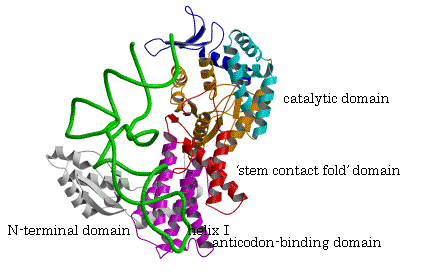
図3. 全長のP.h.ArgRSとtRNAArg(CCU)の複合体の構造
大腸菌由来クラスIaに属するMetRSの生化学的実験とすでに私達が報告した大腸菌とT.t. MetRSの結晶構造からアンチコドンの塩基の結合部位は、アンチコドンドメインのhelix Iとhelix IIIの表面とhelix IIIとhelix IVを繋ぐループ(III -IVループ)の領域であることが示唆された。また、アンチコドンの1番目の塩基のみが異なるIleRSとMetRSのIII -IVループの変異体の実験から、このループがアンチコドンの1番目の塩基と相互作用することが報告された。Met、Ile、Val、Leuは、2番目のアンチコドンがAdeであり、コドンが6種類あり、認識にtRNAのlong variable armの相互作用が指摘されているLeu
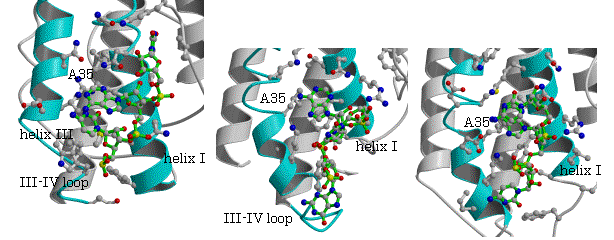
図4.tRNAMet(CAU)、tRNAIle(GAU)と tRNAVal(CAC)のアンチコドンの結合の比較
MetRS (Auifex aeolicusy) IleRS (S.aureus)と T.t. ValRSと
tRNAMet(CAU)の複合体 tRNAIle(GAU)の複合体 tRNAVal(CAC)の複合体
Nature Structural &,
Science 285, 1074 (1999), Cell 103,
793 (2000)
Molecular Biology 10, 931
(2005)
表3.立体構造に基づくhelix Iの一次のアミノ酸配列のアライメント
PhArgRS PDKKIIFRWEDVLNFEGESAPYIQYAHARCSSILRKAEEEGIKVDPETLFKNADFT-KLSE 544 ScArgRS RINNYEFKWERMLSFEGDTGPYLQYAHSRLRSVERNASGITQEKWIN-----ADFSLLKEP
522 ThArgRS PKKQIDFRYQEALSFEGDTGPYVQYAHARAHSILRKAGEWGA----------PDLS-QATP 500 ThMetRS GQDTPVSEEALRTRYEADLADDLGNLVQRTRAMLFRFAEGRIPEPVAG
378 EcMetRS IDDIDLNLEDFVQRVNADIVNKVVNLASRNAGFINKRFDGVLASELAD
414 AaMetRS GQDGDFSKKAILNRINGELANEIGNLYSRVVNMAHKFLGGEVSGARDEE
389 ThIleRS PEADRRFGPNLVRETVRDYFLTLWNVYSFFVTYANLDRPDLKNPPPPEKRPE 674 SaIleRS DYLADVRISDEILKQTSDDYRKIRNTLRFMLGNINDFNPDTDSIPESELL
675 ThValRS TGGQDIRLDLRWLEMARNFANKLYNAARFVLLSREGFQAKEDTPT
620
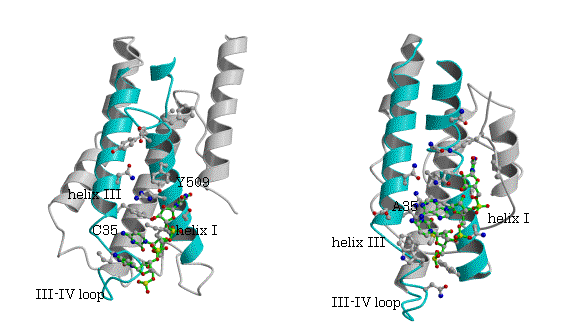
図5. P.h.ArgRSとtRNAArg
(CCU) MetRSとtRNAMet (CAU)
を除いて、MetRS、IleRS、ValRSに共通でそれぞれの種でも保存されているアミノ酸残基を求めたところ、helix I上の表面に存在するAsnが共通に保存されており、MetRS (Aquifex aeolicus)とtRNAMet(CAU)、IleRS (S.aureus)とtRNAIle(GAU)、T.t. ValRSとtRNAVal(CAC)のそれぞれの複合体で、2番目のアンチコドンAdeがhelix I上の保存されたAsnとArg/Pheの間に存在し、Asnの側鎖と水素結合は必ずしも観測されないが、ほぼ予想された位置を占める(図4)。
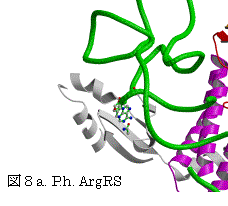
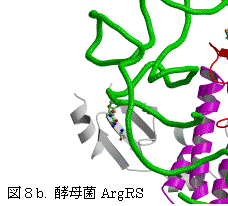
一方、ArgRSでは、対応する位置にTyrが保存されている(表3、図5)。P.h. ArgRSにおいて、2番目のC35がhelix III上のTyr587とスタックし、塩基がIII-IVループの骨格のCOとNHと水素結合を形成し、helix I上の保存されたTyr509から遠く離れている。更に、選択に関与する1番目のアンチコドンC34は、2番目C35の塩基の環と重なり、外を向きタンパク質との相互作用は見られない。3番目のアンチコドンU36 の塩基は、helix IとC末端ループの間の空間に存在するのみで、特別な相互作用は見られない。明らかにこのようにアンチコドンの塩基が位置するとは考えられない。すでに報告された酵母菌 ArgRSとtRNAArg(ICG)でも同様に、2番目のCがhelix III上のTyr565とスタックし、塩基がIII-IVループの骨格のCOとNHと水素結合を形成し、予想されたhelix I上のTyr491
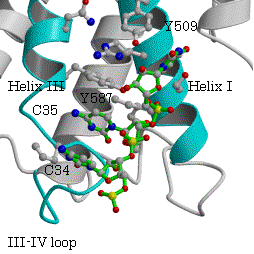
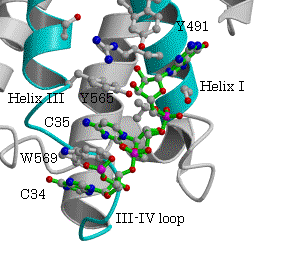
図6.P.h.ArgRSとtRNAArg(CCU) 酵母菌ArgRSとtRNAArg(ICG)
EMB J. 19, 5599 (2000)
から遠く離れている。この結果は、2番目のアンチコドンの塩基の結合サイトについて統一的な説明が出来ない。更に、酵母菌tRNAArg(ICG)では、1番目のIの塩基の環が2番目のCと、III-IVループの酵母菌特有のTrp569の芳香環を挟んでスタックしている。MetRSでは、対応する位置にあるTrpに対し1番目のCytの位置は、Trpの芳香環に対しtRNAArg(ICG)の1番目のIとは逆側すなわち2番目のCの側にある。
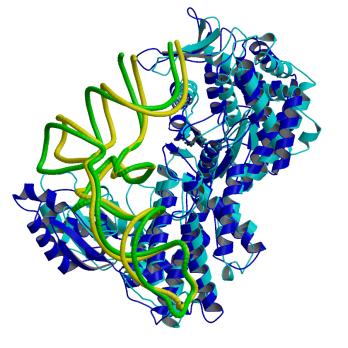
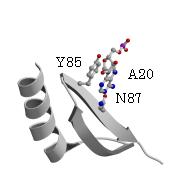
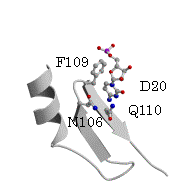
ArgRSのアンチコドンの塩基の結合位置が予想から大きくずれている要因として、ArgRS特有の付加的N末端ドメインとtRNA間の結晶化における相互作用がある。この全長のP.h. ArgRSとtRNAの複合体の構造において、L字形状の屈曲部とN末端ドメインとの相互作用すなわち、DループのA20とN末端ドメインのS3ストランド上のAsn87間の水素結合とA20の塩基とS3上のTyr85のスタッキング型の相互作用が見られた。酵母菌由来ArgRSとtRNAの複合体においても、同様に屈曲部とN末端ドメインの間で相互作用が見られる。他方、酵母菌ArgRSにおいて、これらのN106とQ110のアミノ酸残基の変異体のアミノアシル化反応実験は、tRNAに対する親和性が野生型と変らないこと、すなわちこれらの相互作用がないことを示した。更に、P.h.のArg のコドンのusage が99%を占めているAGAとAGGに対応するtRNA(UCU)とtRNA(CCU)のDループは、前者は9個
図7. P.h. ArgRS (blue) と tRNAArg (green)の複合体と 酵母菌ArgRS
(cyan) と tRNAArg
(yellow)の複合体を重ね合わせた図


|
Codon Usage |
D loop |
anticodon |
|
AGA 19 |
AGC AGGATA |
CUUCUAA |
|
AGG 34 |
AGCCAGGACA |
CUCCUAA |
の塩基、後者は10 個の塩基で構成されており、4番目が1つ欠けたtRNA(UCU)とtRNA(CCU)とにおいて20番目のAdeが同じ配向を取る可能性は低いと思われる。
最初にtRNAArgのアンチコドンがArgRSに結合した後、結晶化においてDループとの相互作用により、本来の位置から動いたとすると予想との違いが説明できる。tRNAArgのアンチコドンが最初にどこと結合するかを明らかにするため、次に、N末端ドメイン欠損型ArgRSとtRNAArgの複合体の立体構造解析を計画した。N末端ドメイン欠損型ArgRSとしてDループとN末端ドメインとの相互作用すべて除くために、S3からN末端側を除くこととし、また、組換え発現において、蛋白質フォールディングに影響を及ぼさないことも考慮し、98番目を切断箇所とした。欠損型ArgRS遺伝子をpET28cに組み込んだ発現ベクターを構築し、大腸菌BL21(DE3)codon+でN末にHis-tag付加したタンパク質の大量発現、精製は確立した。
その他、P.holikoshii由来IleRSはコントラストの作成が困難であったため、P.h.とほぼ同じコドンの使用頻度を示す古細菌Metthanococcus jannashii由来全長IleRS(1050残基)について進めた。N末端にHisTag 付きのコンストラクトを作成しpET28cに組み込んだ発現ベクターを構築し、大腸菌XL1-blueに形質転換しプラスミドの抽出・精製を行った。このプラスミドの発現は検討中である。アンチコドン結合ドメインとtRNAのアンチコドンとの結合を比較するためには、アンチコドン結合ドメインのC末端側にあるドメインは、アンチコドンの最初の結合に関与しないと予想し、このC末端側ドメイン欠損型IleRS(790残基)についても研究を進めた。欠損型IleRS遺伝子をpET28cに組み込んだ発現ベクターを構築し、大腸菌BL21(DE3)codon+で大量発現させ、Ni-NTA Superflowカラム、Heparinカラムを通して精製した。現在、結晶化の条件検討を行っている。P.h. GlyRSとP.h. TrpRSについても同様にpET28cに組み込んだ発現ベクターを構築し、BL21(DE3)codon+で大量発現し精製を行った。これらのタンパク質も結晶化を試みているが、現在のところP.h. TrpRSについては、得られた結晶の強度測定を行ったが、低分解能であった。今後、tRNAとの複合体の構造解析も進める予定である。
2)BirAとbiotin複合体の構造解析の結果
Biotin Protein ligase (BirA)は、ATPを補酵素としMg2+イオン存在下でビオチン酵素のLysにBiotinを付加する反応を触媒する(式1)。この酵素反応は、aaRSにおいてATPを補酵素としMg2+イオン存在下でtRNAにアミノ酸を付加する反応に対応する(式2)。
Biotin + apoprotein + ATP → biotinyl-holoprotein + AMP + PPi (式1)
BirA, Mg2+
amino acid + tRNA + ATP → aminoacyl-tRNA + AMP + PPi (式2)
aaRS, Mg2+
更に、E.coliのBirA(PDB:1BIA)の構造が、aaRSのクラスIIの触媒ドメインと同じ逆平行b-シートから成ることが示された。クラスIIに属するaaRSとThermus thermophilus(T.t.)由来BirA(297残基)の構造の比較から、ATP とMg2+イオンのタンパク質への配位の共通性を見出すためにT.t.由来BirAとBiotinの複合体及びBirA単独の立体構造を決めた。反射強度は、Spring8及びPFで収集した。すでに解析されたE. coli由来BirA (PDB:1BIA)をサーチモデルとして分子置換法を行ったが解が得られなかった。V27M、L210M、L251Mの3残基を置換したmutantを作成し、SeMetタンパク質を用いてMAD測定を行い、プログラムSOLVE/RESOLVEで位相を求め、電子密度図からE. coli由来BirAのモデルを参考に分子構造を構築した。現在のところBirA とBiotinの複合体は、分解能2.35 Åで、Rfactor=0.235(Rfree=0.294)である。単独のBirAについては、2種類の結晶、結晶I(tetra)(tetrahedral、P41211、分解能3.1 Å)と結晶II(ortho)(orthorhombic、P212121、分解能3.0 Å)が得られた。 現在、結晶I(tetra)は、Rfactor=0.236(Rfree=0.356)、結晶II(ortho) は、Rfactor=0.259(Rfree=0.363)である。
T.h. BirAとBiotinの複合体の構造を図9に示す。T.h. BirAは、helix-turn-helix
motifを含むN末端ドメイン、7本の逆平行bストランドから成るb-シート構造をもつ中央の触媒ドメイン、C末端ドメインから成る。Biotinの結合したBirA と単独の結晶I(tetra)、結晶II(ortho)のBirAのCaを重ねると各ドメインの構造は、保存されているが、全体としてドメイン間の相対的配向がずれている。N末端ドメインと触媒ドメインは、残基57-74にわたる長いヒンジで繋がれている。C末端ドメインと触媒ドメイン間の相互作用は、多くの種
Catalytic
domain N-terminal
domain C-terminal
domain
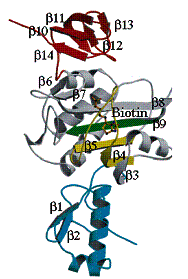
図9.T.h. BirAとBiotinの複合体のリボンモデル
で保存されているL279とW163 の側鎖の間に見られる。興味あることに、Biotinが結合したBirA と単独のBirAの結晶I(tetra)では、これらのドメイン間で同じ配向を取るが、単独のBirA結晶II(ortho) は大きくずれている。W163はb6-b7のターン(162-KWPND-
166)に含まれ、このターンの配向が前者とBirA結晶II(ortho)で異なる。
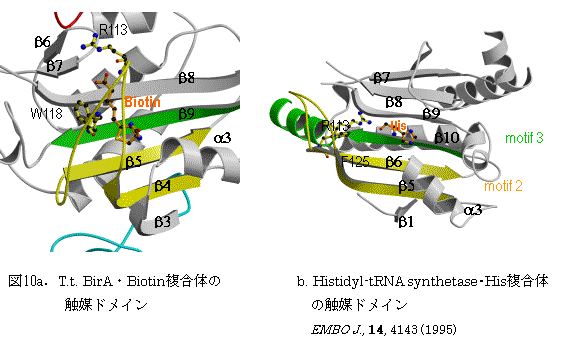
BirAの触媒ドメインは、クラスIIのaaRSと同じトポロジーを持つ。図10にBirA の触媒ドメインとクラスIIに属するhistidyl-tRNA
synthetase(PDB:1HTT)の触媒ドメインの活性部位の比較を示す。histidyl-tRNA synthetaseの活性部位であるモチーフ2とモチーフ3に対応して、BirAではそれぞれb4とb5とその間のループとb9がある。ATPのアデニン塩基は、aaRSでは、モチーフ2のループ、BirAでは、b4-b5間ループ近傍にある。Biotinのウレイド環は、b-シ−トのb8、b9 とb4-b5の間のループに挟まれている。b8の174-KLAGVLIE-181のKGLEとb9の194-LGVGVNV-200のGGNは分子の表面にあり、多くの種で保存されている。b4-b5間ループは、GRリッチな配列110-GRGRRGR-116を含んでおり、ほとんどの種で保存されている。BirA単独の結晶では、111から118残基の領域では乱れがあり、電子密度が観測されない。すべての種に保存されているb8のLys174は、Biotinの吉草酸のカルボキシル基を攻撃可能な位置にあるにも係わらずBiotinはこのLys残基には転移されない。ATPのリン酸基は、b6-b7のターン近傍に存在すると思われる。このb6-b7のターンの配列162-KWPND-166は、P164以外調べられた全ての種において保存されている。今後、これまで解析されたクラスIIのaaRSとの立体構造の比較を行い、基質Biotinとアミノ酸、ATPの配向についてその共通の機構を検討する。
構造解析の過程にあるタンパク質の数及び状況
|
|
タンパク質の名前 |
発現・精製 |
結晶化 |
分解能 |
構造解析 |
|
1 |
P.h. ArgRSとP.h. tRNAの複合体 |
○ |
○ |
2.0 Å |
解析終了 |
|
2 |
P.h. ArgRS、P.h. tRNAArg(CCU)とATP-PNPの複合体 |
○ |
○ |
2.0 Å |
解析終了 |
|
3 |
P.h. GlyRS |
○ |
△ |
|
|
|
4 |
P.h. TrpRS |
○ |
△ |
10 Å |
|
|
5 |
M.j. IleRS |
○ |
△ |
|
|
|
6 |
T.t. BirAとbiotinの複合体 |
○ |
○ |
2.35Å |
ほぼ解析終了 |
|
7 |
T.t. BirA単独 |
○ |
○ |
3.1 Å |
解析中 |
・ ArgRS:アルギニル-tRNA合成酵素、GlyRS:グリシル-tRNA合成酵素、TrpRS:トリプトファニル-tRNA合成酵素、IleRS:イソロイシル-tRNA合成酵素
・ T.t:Thermus termophilus、P.h:Pyrococcus horikoshii、M.j.:Metthanococcus jannashii
・○:成功、△:検討中
PDBへの登録状況4
・ PDB Entry No 1vdn:酵母菌サイクロフィリンAとtetrapeptideの複合体の結晶構造
・ PDB Entry No 1w0y:高度好熱菌由来メチオニル-tRNA合成酵素Y225F変異体(isopropanol)の結晶構造
・
PDB Entry No 2d5b:高度好熱菌由来メチオニル-tRNA合成酵素Y225F変異体(PEG6000)の結晶構造
・
PDB Entry No 2d54:高度好熱菌由来メチオニル-tRNA合成酵素Y225A変異体の結晶構造
